Bayesian
Estimation of Species Divergence Times Integrating Fossil and
Molecular Information
Because genes
accumulate changes over time at a constant rate, the genetic
distance between two species, measured by the number of changes
accumulated, will be proportional to the time of species
divergence. Thus molecules can serve as a clock, keeping time of
species divergence by the accumulated changes. If fossil records
or geological events can be used to assign an absolute geological
time to a species divergence event on the phylogenetic tree, one
can convert all calculated genetic distances into absolute
geological times. This rationale for molecular clock dating has
recently been extended to deal with local variation in
evolutionary rate. Critical to molecular dating is the use of
fossil information to calibrate the clock. In this project, we are
developing statistical models and computer programs to analyze
fossil and molecular data to accurately represent and incorporate
the information in the fossil record in molecular dating analysis.
The new methods will be applied, for example, to analyze datasets
to estimate divergence times among mammals and to date viral
transmission events. The project is funded by the BBSRC, UK, grant
number BB/J009709/1,
and it will run between March 2012 and February 2015.
The grant holder is Prof. Ziheng Yang (University College London),
who is working in collaboration with Prof. Philip CJ Donoghue
(Bristol University). The named postdoctoral fellow in the grant
is Mario dos Reis (University College London).
The project is now finished and this website will not be
updated any more. These are a few examples of the work that was
carried out during the grant, as well as work done during a previous
related grant:
The uncertainty of divergence time estimates under relaxed clock
models
In a paper published in Systematic Biology (early 2015, read it here) we
study how uncertainty in divergence time estimates decreases with
the amount of molecular sequence data under relaxed clock models.
This study is an extension of our previous work in the Journal of
Systematics and Evolution (here)
where we study similar issues under the strict clock. With the
relaxed clock, uncertainty in divergence time estimates is due to
uncertainty in the fossil calibrations, uncertainty due to limited
sequence data (sampling errors in the data), and uncertainty in
rate variation among lineages and among loci. As the number of
loci in the analysis is increased, the total uncertainty decreases
until it reaches a limiting value determined by the fossil
calibrations. Here the number of loci (each of which has its own
relaxed clock) is the crucial factor in reducing uncertainty.
Thus, analysing many genes (or loci) as separate partitions is
preferable than analysing a super-gene concatenation.
Effect of the rate prior on times estimates with multiple loci
Estimating divergence times using multiple loci (multiple genes or
partitions) requires constructing a joint prior on the substitution
rates across loci. For example, if we are analysing 10 genes, then
we would estimate 10 substitution rates, one for each gene.
Virtually all Bayesian molecular dating programs use i.i.d. priors
for the locus rates, that is, they assume that the substitution
rates at loci are independent, identically distributed random
variables from some distribution. In a paper in Systematic Biology
(early 2014, read it here) we show
that the innocent looking i.i.d. rate prior is problematic, leading
to overtly precise estimates of divergence times, and if the rate
prior is misspecified (say, if it assumes that genes evolve too fast
or too slow compared to the real rate), estimated times will
converge with absolute precision to wrong values. The more loci we
analyse, the worst the results will be. In our paper we implement a
new rate prior based on the Dirichlet distribution that is robust to
prior misspecification and that does not lead to overtly precise
time estimates. The new prior is implemented in MCMCTree v4.8. Users
of MCMCTree will not notice any difference in usage with respect to
older versions, other than posterior time estimates will be more
robust to rate prior misspecification. In our paper we also give a
strategy to construct a safe i.i.d. rate prior for users of other
Bayesian programs (BEAST, MrBayes and so on implement the
problematic i.i.d. prior).
Virus dating
The MCMCTree program for dating now implements models to obtain
divergence times among molecular sequences that have been serially
sampled in time. The most common case is virus dating. The program
implements a new time prior based on the
birth-death-sequential-sampling (BDSS) process. An important finding
is that estimation of deep divergence times (say the root of the
phylogeny) can be very sensitive to the parameters of the BDSS
prior, even with well serially sampled sequences. You can read the
paper by Tanja Stadler and Ziheng Yang published in Systematic
Biology in April 2013 here.
The unbearable uncertainty of Bayesian divergence time
estimation
Bayesian estimation of divergence times of extant species using
molecular data is an unconventional statistical problem. Molecular
data provide information only about the distances among species on a
phylogeny, but not about the geological ages of groups of species
nor the molecular evolutionary rate. Information from the fossil
record is needed to convert molecular distances into divergence time
estimates. This means that as the amount of molecular data is
increased, the uncertainty in estimated divergence times does not
approach zero, but approaches a limiting value determined by the
uncertainty in the fossil calibrations. In this project we extend
the mathematical theory of the uncertainty in divergence time
estimation. In particular, we show that the uncertainty in time
estimates approach their theoretical limit at the rate 1/n, with n to be the sample size (the
number of sites in the molecular sequence alignment). We also
studied the effect of conflicting fossils and sequence data on the
precision of time estimates. You can read the paper here.
The paper was featured in the cover of the
2014 January issue of the Journal of Systematics and Evolution.
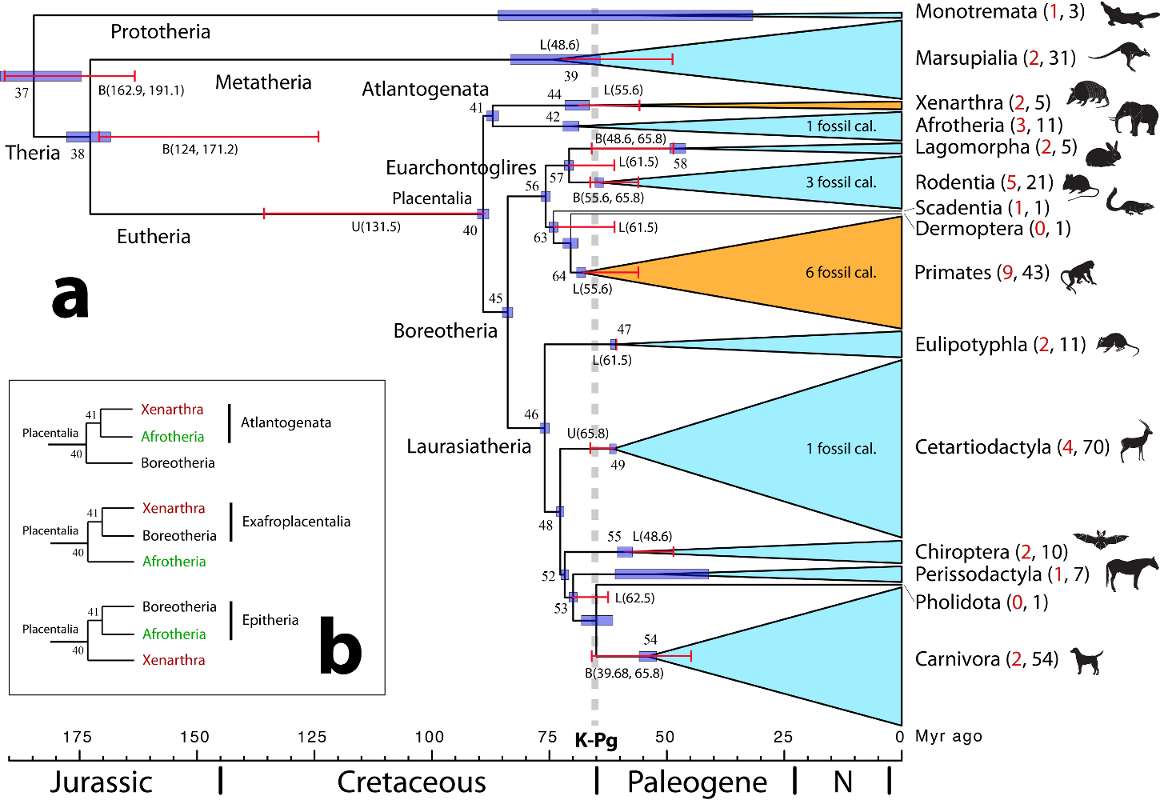
The timescale of mammalian phylogeny
The fossil record shows the sudden appearance of Placental mammals
65 My ago, soon after the Cretaceous-Paleogene mass extinction event
when 76% of species, including non-avian dinosaurs, died out. Some
molecular studies have placed the origin and diversification of
placental mammals deep in the Cretaceous, before the mass extinction
event. On the other hand, palaeontological analysis of the fossil
record indicate a diversification peak of placental orders in a
short 16 My post-Cretaceous window. The discrepancies between
estimates of mammalian divergence between molecular and
palaeontological studies have been considered unacceptably large. We
revised the time-line of mammalian diversification by Bayesian
analysis of a large alignment (21 million sites) from 36 mammalian
genomes, together with mitochondrial data from 274 mammal species.
We used 26 soft, non-limiting fossil constraints to calibrate the
molecular clock. Our analysis indicates that intra-ordinal
diversification of placental mammals occurred in a 20 My
post-Cretaceous window, in accordance with palaeontological
estimates. On the other hand, we find that the last common ancestor
of placentals originated in the Creataceous (90-88 My ago). We find
that genomic data reduces uncertainty in divergence time estimates
towards the theoretical limit of precision and allowed us to
confidently reject a pre-extinction diversification of placentals.
Other famous controversies of mismatch between palaeontological and
molecular time estimates, such as the origin of animal phyla or of
flowering plants, are likely to be resolved with a genomic-scale
approach.
You can read the full paper published in May 2012 in Proceedings of
the Royal Society B here, and a
comment in EvoDevo here. You can
also download a large dated mammal phylogeny (274 species) here. The alignment and
trees with fossil calibrations are here.
Below is a simplified figure that summarises the finding of the
project: nearly all modern crown placental orders postdate the K-Pg
event, in accordance with a rapid diversification of mammals after
the mass extinction.
In early 2013, O'Leary and colleagues (Science, 339: 662)
published an analysis of a large morphological matrix of extant and
fossil mammal species to reconstruct and date the placental mammal
ancestor. They incorrectly treated fossil ages as divergence time
estimates for crown groups. For example, they recognise Protungulatum donnae (giving it
an age of 64.85 My) as the oldest placental mammal fossil. They then
estimate the origin of placentals to be the same as the age of P. donnae, therefore
concluding, wrongly, that Placentalia originated 64.85 My ago after
the K-Pg extinction. The molecular data is not compatible with such
a young age for Placentalia. Such overtly simplistic interpretation
of the fossil record is naive and misleading, and ignores the
integrative work that paleontologists and molecular biologists have
carried out on the theory of divergence time estimation during the
past two decades. We published a rebuttal in Biology Letters in
January 2013 here.
Our critique was discussed by Nature here, by
The Scientist here
and by Discovery News here.
Approximate likelihood calculation for Bayesian estimation of
species divergence times
Bayesian estimation of divergence times from molecular data is
computationally expensive. The posterior distribution of divergence
times cannot be calculated analytically, so we must rely on Markov
Chain Monte Carlo (MCMC) methods to obtain an approximation to the
posterior distribution. Calculation of the log-likelihood of a
molecular sequence alignment during MCMC is the most expensive part
of the posterior computation. Thorne and colleagues (1998, MBE
15:1647) proposed a normal approximation to the log-likelihood
function to speed up computation during the MCMC. However, the
accuracy of this approximation was never fully tested. In this
project we performed a detailed mathematical analysis of the
approximation and suggested using variable transforms to increase
accuracy. Extensive testing using real molecular data indicates that
the approximation is highly reliable. Our new ARCSINE based
transform is now the default approximate likelihood method in the
program MCMCTREE. You can read the paper, published in MBE in
February 2011, about the approximate likelihood method here. With the
traditional exact method Bayesian estimation of the divergence times
of a few species may take from a few days to months, while with the
approximate method analysis in one to two days are possible. The
exact method becomes prohibitively expensive for genome scale data.
The approximate method in MCMCTREE allowed us to estimate the
divergence times of mammal species using and alignment of over 21
million sites, and solve a long standing controversy in molecular
evolution (see above and here).
The impact of fossil calibrations on divergence time estimation
The construction of a multidimensional prior in Bayesian statistics
is a complicated problem. Bayesian estimation of species divergence
times involves the construction of the joint prior for the
divergence times. For a set of s
species, there are s-1
times to be estimated, and an s-1
multidimensional joint time prior must be constructed. Different
Bayesian divergence time estimation programs (such as MULTIDIVTIME,
BEAST or MCMCTREE) use different strategies to integrate fossil
information into a phylogeny in order to construct the time prior.
These different strategies may lead to different priors from
seemingly similar fossil calibration densities. In turn, the
different priors may lead to surprisingly different posterior time
estimates for the different programs, despite the use of the same
molecular data and fossil information. In this project a detailed
mathematical analysis of the construction of the time prior in
MCMCTREE and MULTIDIVTIME was made. This work highlights the
critical importance of calculating the time prior explicitly and
comparing it to the fossil calibration densities. You can read the
full paper in Syst. Biol. (first published in Nov 2009) by Jun Inoue
et al. here. (Also,
you may want to look at fig. 3 in our mammal paper for an
example comparing the marginal time prior and the fossil calibration
densities).
For any questions about this web-page contact Mario dos Reis or
Ziheng Yang.
Last updated February 2015.