Research
Our research concentrates on computational biology, including the areas of molecular evolution, molecular systematics, population genetics, and phylogenetics/phylogenomics.
We develop statistical models of DNA or protein evolution to be used in reconstructing species phylogenies and in understanding the mechanisms of molecular sequence evolution. We make extensive use of statistical methods, such as maximum likelihood and Bayesian theory, and computer simulation as well.
Real data analysis is another major undertaking in the group, with detection of adaptive molecular evolution and comparative genomics as the focus. Real data analysis serves as the motivation for the more theoretical work.
The following topics cover the directions of our current research:
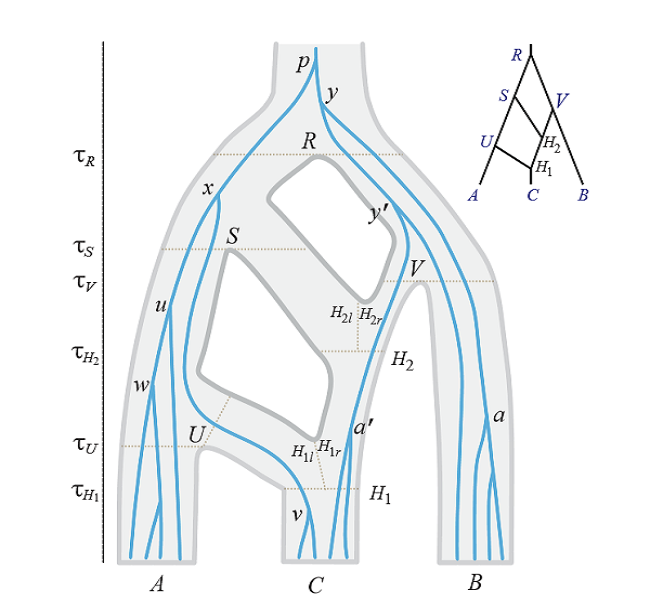
-Bayesian estimation of phylogenies and divergence dates. We develop Markov chain Monte Carlo (MCMC) algorithms to estimate molecular phylogenies and species divergence dates under Bayesian framework. Details about our research in this area can be found here.
-Markov models of codon substitution and detection of adaptive molecular evolution. This involves development of models of codon substitution for comparative analysis of protein-coding DNA sequences. The nonsynonymous/synonymous substitution rate ratio provides a measure of selective pressure on the protein, which can be used to detect positive Darwinian selection acting on the protein sequence.
-MCMC algorithms under multispecies coalescent models. We produce MCMC algorithms for analysing population samples from multiple loci with different data types (such as sequences and microsatellites), accounting for population demographics, migration/introgression, and relaxed molecular clock.
-Comparative analyses of viral and mammalian genomes. This project intends to perform genome-wide comparisons to identify changes in selective pressures after gene duplications or species divergence, or after viral transmission to a new host.
{kind=link}